- Gen 10
- Gen 99
- Gen 383
- Gen 444
Descripción de la actividad
La tarea consiste en llevar a cabo un Análisis de la Varianza Univariante (ANOVA) y Multivariante (MANOVA) sobre el conjunto de datos “geneData” del paquete “Biobase”. Esta tabla contiene los valores de expresión de 500 genes medidos sobre 26 pacientes. El ANOVA se llevará a cabo sobre los genes correspondientes a las filas 10, 99, 383 y 444 de la tabla de datos “geneData”. Los grupos se definirán por el factor “tipo de cáncer” según los datos de la tabla “tipos_cancer.xlsx”. El alumno aplicará el tipo de ANOVA más adecuado a cada variable dependiente, en función de la normalidad y la homocedasticidad. El MANOVA se llevará a cabo sobre los mismos grupos que en el ANOVA y tomando las mismas variables dependientes, pero analizándolas conjuntamente. En el caso de que sea necesario, llevar a cabo los contrastes post-hoc pertinentes.
Datos
# Instalar Biobase si no está instalado
if (!require("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("Biobase")
# Cargar la librería
library(Biobase)
# Cargar los datos geneData
data("geneData")
# Mostrar las primeras filas y columnas de la tabla
head(geneData)
# Ver los datos completos
View(geneData)
# Asignamos tipo de cáncer (factor) a cada uno de los 26 individuos:
cancer <- factor(c(1,3,2,2,2,2,2,1,2,3,3,2,2,1,2,1,1,3,1,1,3,2,1,3,3,3),
levels = 1:3,
labels = c('colon','lung','liver'))
Gen 10 - Normalidad
Código
gen10 <- geneData[10, ]
means <- as.numeric(by(gen10, cancer, mean))
print(means)
layout(matrix(1:2, ncol = 2)); cols=c('blue','red','green','black')
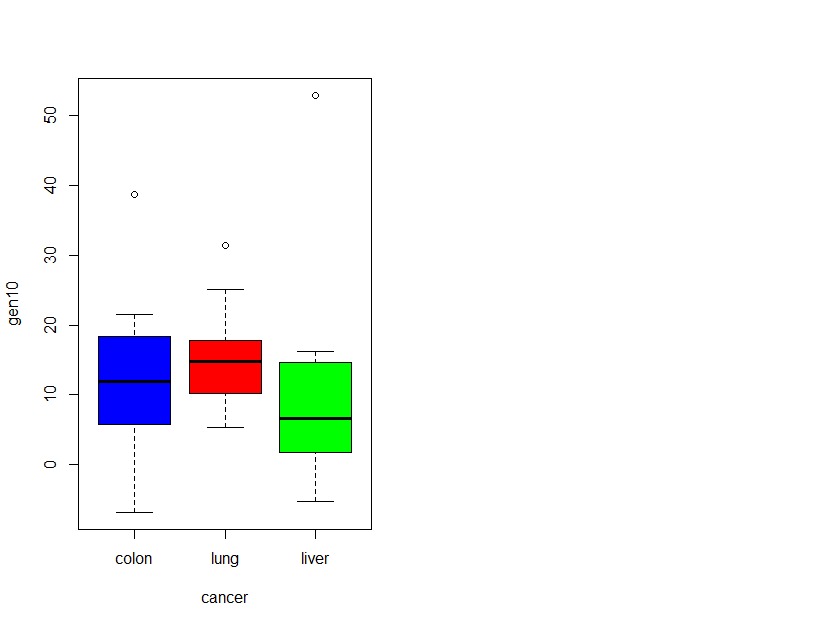
boxplot(gen10 ~ cancer, col=cols[1:3])
by(gen10, INDICES = cancer, FUN = function(x) shapiro.test(x)$p.value)
Boxplot
Resultados
Medias: colon (13.01), lung (15.93), liver (11.68)
Normalidad: colon (p=0.702), lung (p=0.531), liver (p=0.020) → liver no cumple normalidad
Gen 10 - Homocedasticidad
Código
library(car)
leveneTest(gen10, cancer, center = mean)
Resultados
Dado que p=0.365 > 0.05, se cumple homocedasticidad.
Gen 10 - ANOVA
Código
anova_10 <- kruskal.test(gen10 ~ cancer)
print(anova_10)
Resultados
Kruskal-Wallis p=0.2599 → No hay diferencias significativas
Gen 99 - Normalidad
Código
gen99 <- geneData[99, ]
means <- as.numeric(by(gen99, cancer, mean))
print(means)
layout(matrix(1:2, ncol = 2)); cols=c('blue','red','green','black')
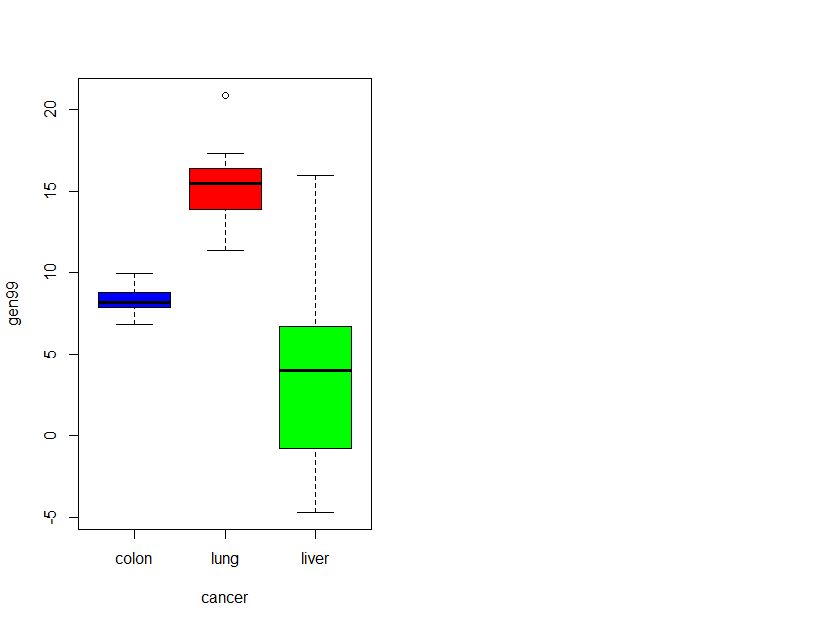
boxplot(gen99 ~ cancer, col=cols[1:3])
by(gen99, INDICES = cancer, FUN = function(x) shapiro.test(x)$p.value)
Boxplot
Resultados
Medias: colon (8.31), lung (15.26), liver (3.90)
Normalidad: p > 0.05 en todos los grupos → se cumple normalidad
Gen 99 - Homocedasticidad
Código
levene_res_99 <- leveneTest(gen99,cancer, center = mean)
print(levene_res_99)
Resultados
Como p=0.02893 < 0.05, no se cumple la homocedasticidad.
Gen 99 - ANOVA
Código
anova_99 <- oneway.test(gen99 ~ cancer)
print(anova_99)
pairwise.t.test(gen99, cancer, p.adjust = "fdr")
Resultados
p=3.127e-05 → hay diferencias significativas
Post-hoc: todas las comparaciones significativas
Gen 383 - Normalidad
Código
gen383 <- geneData[383, ]
means <- as.numeric(by(gen383, cancer, mean))
print(means)
layout(matrix(1:2, ncol = 2)); cols=c('blue','red','green','black')
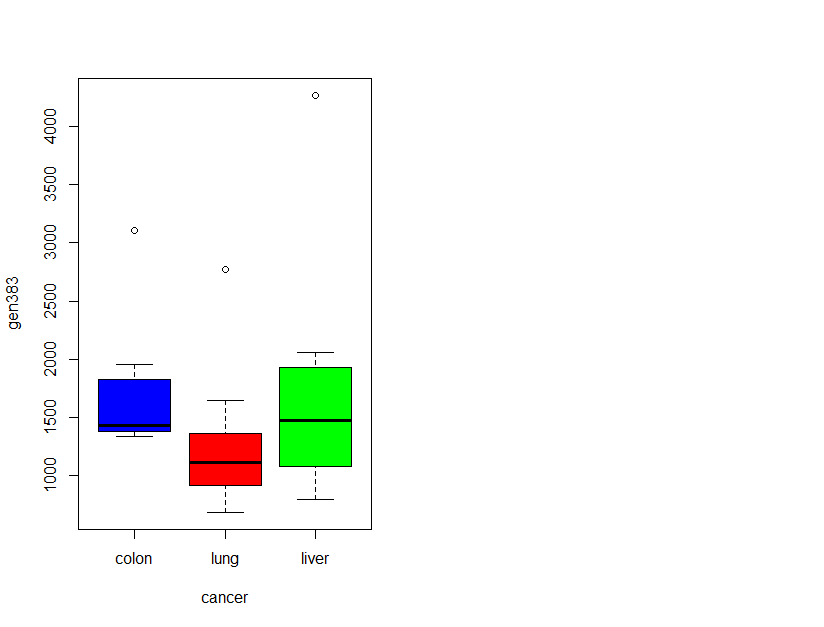
boxplot(gen383 ~ cancer, col=cols[1:3])
by(gen383, INDICES = cancer, FUN = function(x) shapiro.test(x)$p.value)
Boxplot
Resultados
Medias: colon (1714.91), lung (1267.76), liver (1754.59)
Normalidad: p < 0.05 en todos los grupos → no se cumple normalidad
Gen 383 - Homocedasticidad
Código
levene_res_383 <- leveneTest(gen383,cancer, center = mean)
print(levene_res_383)
Resultados
Como p=0.4239 > 0.05, se cumple homocedasticidad.
Gen 383 - ANOVA
Código
anova_383 <- kruskal.test(gen383 ~ cancer)
print(anova_383)
Resultados
Kruskal-Wallis p=0.06969 → no hay diferencias significativas
Gen 444 - Normalidad
Código
gen444 <- geneData[444, ]
means <- as.numeric(by(gen444, cancer, mean))
print(means)
layout(matrix(1:2, ncol = 2)); cols=c('blue','red','green','black')
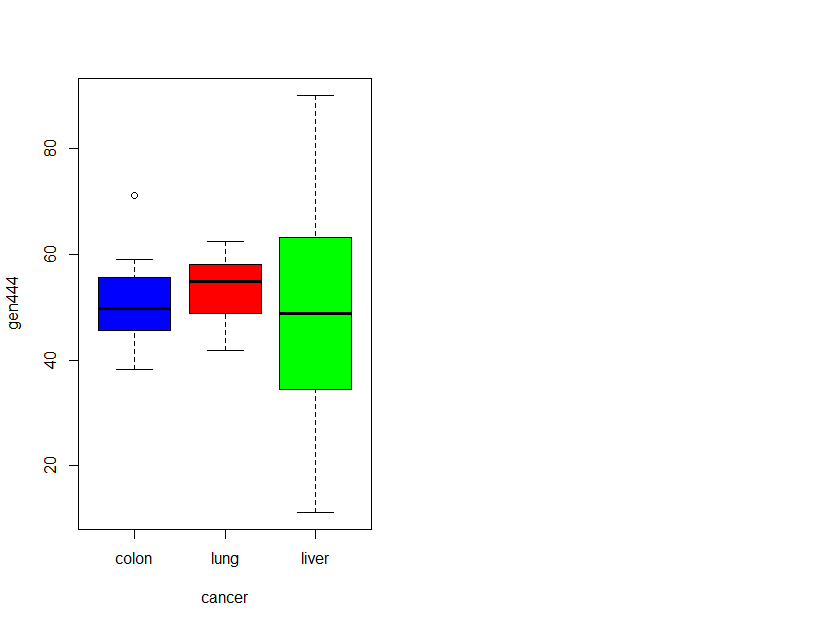
boxplot(gen444 ~ cancer, col=cols[1:3])
by(gen444, INDICES = cancer, FUN = function(x) shapiro.test(x)$p.value)
Boxplot
Resultados
Medias: colon (51.40), lung (53.60), liver (49.25)
Normalidad: p > 0.05 en todos → se cumple normalidad
Gen 444 - Homocedasticidad
Código
levene_res_444 <- leveneTest(gen444,cancer, center = mean)
print(levene_res_444)
Resultados
Como p=0.01587 < 0.05, no se cumple homocedasticidad.
Gen 444 - ANOVA
Código
anova_444 <- oneway.test(gen444 ~ cancer)
print(anova_444)
Resultados
p=0.8044 → no hay diferencias significativas
MANOVA
# Comprobación de normalidad multivariante
normalidad_multivariante <- mvn(data = genes_seleccionados[1:4], mvnTest = "mardia")
# Homocedasticidad multivariante
homocedasticidad_multivariante <- boxM(as.matrix(genes_seleccionados[1:4]), genes_seleccionados$CancerType)
# MANOVA
modelo_manova <- manova(cbind(Gen_10, Gen_99, Gen_383, Gen_444) ~ CancerType, data = genes_seleccionados)
summary(modelo_manova)## Df Pillai approx F num Df den Df Pr(>F)
## CancerType 2 0.63804 2.4595 8 42 0.02778 *
## Residuals 23
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1# Contrastes post-hoc
summary.aov(modelo_manova)## Response Gen_10 :
## Df Sum Sq Mean Sq F value Pr(>F)
## CancerType 2 86.0 42.982 0.2435 0.7859
## Residuals 23 4060.1 176.528
##
## Response Gen_99 :
## Df Sum Sq Mean Sq F value Pr(>F)
## CancerType 2 593.21 296.605 18.127 1.878e-05 ***
## Residuals 23 376.35 16.363
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Response Gen_383 :
## Df Sum Sq Mean Sq F value Pr(>F)
## CancerType 2 1348323 674161 1.1025 0.3489
## Residuals 23 14063592 611461
##
## Response Gen_444 :
## Df Sum Sq Mean Sq F value Pr(>F)
## CancerType 2 84.4 42.22 0.1867 0.8309
## Residuals 23 5200.3 226.10# Realizar MANOVA
modelo_manova <- manova(cbind(Gen_10, Gen_99, Gen_383, Gen_444) ~ CancerType, data = genes_seleccionados)
# Resumen MANOVA
res_manova <- summary(modelo_manova)
# Extraer y formatear los resultados de MANOVA
manova_resumen <- as.data.frame(res_manova$stats)
colnames(manova_resumen) <- c("Statistic", "Pillai", "Wilks", "Hotelling-Lawley", "Roy", "Pr(>F)")
# Mostrar tabla en formato legible
library(knitr)
kable(manova_resumen, caption = "Resultados del MANOVA")| Statistic | Pillai | Wilks | Hotelling-Lawley | Roy | Pr(>F) | |
|---|---|---|---|---|---|---|
| CancerType | 2 | 0.6380393 | 2.459473 | 8 | 42 | 0.0277841 |
| Residuals | 23 | NA | NA | NA | NA | NA |
Conclusion Manova
Conclusiones:
- Pruebas de Normalidad:
- Gen_10 y Gen_383: No cumplen normalidad → Kruskal-Wallis.
- Gen_99 y Gen_444: Cumplen normalidad, pero violan homocedasticidad → Welch ANOVA.
- Pruebas ANOVA/Kruskal-Wallis:
- Gen_99 mostró diferencias significativas entre grupos (p-value < 0.05).
- Otros genes no mostraron diferencias significativas.
- MANOVA:
- El análisis multivariante indica diferencias conjuntas entre genes según el tipo de cáncer (p = 0.027).
Resultados y visualizacion
# Visualizar diferencias entre grupos para un gen como ejemplo
ggplot(genes_seleccionados, aes(x = CancerType, y = Gen_10)) +
geom_boxplot() +
theme_minimal() +
labs(title = "Expresión del Gen 10 por tipo de cáncer", x = "Tipo de Cáncer", y = "Expresión del Gen 10")
Conclusiones del análisis
Pruebas de Normalidad:
• Algunos genes cumplen con la normalidad, mientras que otros no. Esto llevó a usar diferentes pruebas estadísticas según los resultados (ANOVA estándar o alternativas no paramétricas).
• Gen_10 y Gen_383 no cumplen normalidad, lo que implica usar pruebas no paramétricas como Kruskal-Wallis.Pruebas de Homocedasticidad:
• Los resultados del test de Levene muestran que hay homocedasticidad para algunos genes pero no para otros.
• Gen_99 y Gen_444 violan la homocedasticidad, lo que justifica el uso de Welch ANOVA en lugar de ANOVA estándar.MANOVA:
• El análisis MANOVA fue correctamente aplicado considerando los genes seleccionados como variables dependientes y el tipo de cáncer como el factor independiente.
• Los resultados permiten identificar diferencias entre los tipos de cáncer al analizar las expresiones génicas de forma conjunta.